
Quantum Chemical Insights into the Electronic, Vibrational and Thermodynamic Properties of Chloro-Substituted Anisole
Keywords:
Anisole , Density functional theory , Electronic structure , Vibrational analysis , Thermodynamic parametersAbstract
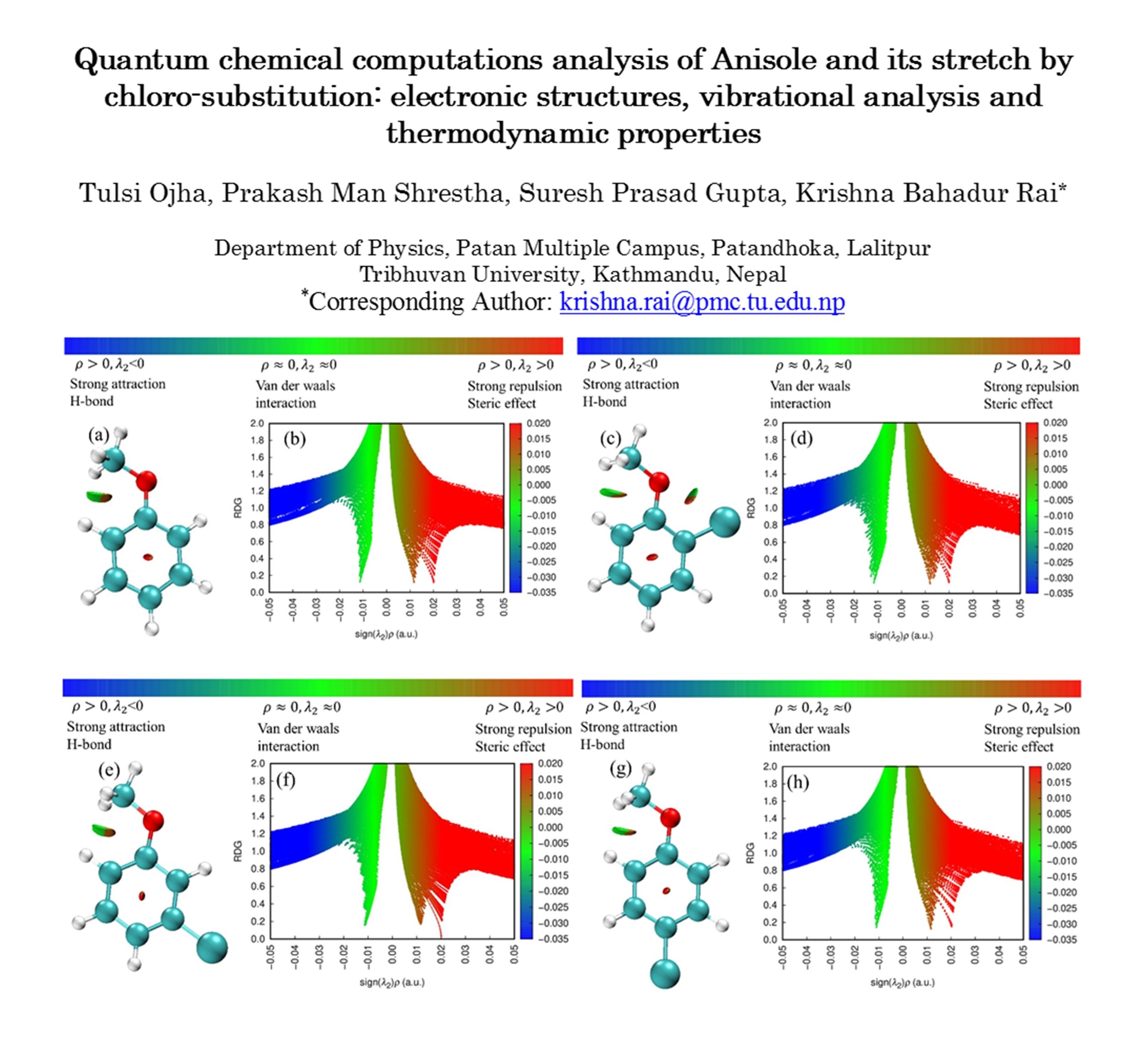
Anisole, also known as methoxybenzene, is an organic compound with the formula \(CH_{3}OC_{6}H_{5}\). It is a colorless liquid with a sweet, aromatic odor similar to anise seeds and is used as an intermediate in the synthesis of pharmaceuticals, agro-chemicals, and fragrances. This study is about the quantum chemical insight into the Electronic, vibrational, and thermodynamic properties. The study first analyzed the optimized molecular structures and identified different physical properties. The Non-Co-valent Interactions (NCI)-Reduced Density Gradient (RDG) reveal weak hydrogen bonding and strong repulsive interactions. Molecular electrostatic potential mapping shows higher reactivity in chlorine-substituted anisole, with key reactive sites at oxygen, chlorine, and carbon atoms. Anisole has an excitation energy of 5.770 eV and a lower electronegativity of 3.2850 eV, indicating kinetic stability. Para-chloroanisole (PCA) exhibits the lowest energy gap at 5.481 eV and chemical hardness of 2.7405 eV, suggesting high reactivity. The quantum energy levels are observed in the density of States (DOS) spectrum. Due to their electronegative nature, chlorine atoms show positive charges while oxygen atoms, owing to their electron-withdrawing properties, show negative Mulliken charges, respectively, and serve as key reactive sites. In addition to that, Infrared (IR), Raman, and UV-visible spectra reveal different vibration modes and spectral intensities. Thermodynamic functions such as constant pressure (Cp), enthalpy (H), and entropy (S) for all studied molecules increase with temperature.
References
[1] Dionisio, K.; Phillips, K.; Price, P.; “The Chemical and Products Database, a resource for exposure-relevant data on chemicals in consumer products”. Sci Data., 5: 180125, 2018.
[2] Vani, B.; Pabba, M.; Kalyani, S., Kalyani, S.; Sridhar, S.; “Separation of Anisole and Valuable Byproducts from Liquid Reaction Mixtures by Solvent Extraction and Multicomponent Distillation”. J. Solution Chem., 50: 160–177, 2021.
[3] El-Saady, A. A.; Roushdy, N.; Farag, A. A. M.; “Exploring the molecular spectroscopic and electronic characterization of nanocrystalline Metal-free phthalocyanine: A DFT investigation”. Opt. Quantum Electron., 55, 662, 2023.
[4] Ayachi, S.; Bergaoui, S.; Ben Khalifa, I.; Haj Said, A.; Chemek, M.; “On the photo-physical properties of soluble oligomer from anodic oxidation of chlorine-substituted anisole (OPClAn)”. Synth. Met., 166: 22, 2013.
[5] Malongwe, J.; K. E., Nachtigallová, D.; Corrochano, P.; and Klán, P.; “Spectroscopic Properties of Anisole at the Air–Ice Interface: A Combined Experimental–Computational Approach”. Langmuir, 32(23): 5755-5764, 2016.
[6] Ali, M.; Mansha, A.; Asim, S.; Zahid, M.; Usman M., et al.; “DFT Study for the Spectroscopic and Structural Analysis of p-Dimethylaminoazobenzene”. J. Spectroscopy, 2018: 1-15, 2018.
[7] Shastri, A.; Das, A. K.; Rajasekhar, B. N.; “The electronic absorption spectrum of Anisole was studied by photo absorption spectroscopy and quantum chemical calculations”. J. Quant. Spectrosc. Radiat Transfer, 242: 2020.
[8] Frisch, M.J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; et al.; Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009.
[9] Dennington, R.; Keith, T. A.; Millam, J. M.; GaussView, version 6.0. 16., Semichem Inc, Shawnee Mission, 2016.
[10] Jana, S.; Samal, P.; “Assessment and Benchmarking of Proposed Exchange-Correlation Approximations in Density Functional Theory”. Doctoral dissertation, School of Physical Sciences, NISER, Bhubaneswar, 2021.
[11] Nagy, B.; Jensen, F.; “Basis sets in quantum chemistry”. Rev. Comput. Chem., 30: 93-149, 2017
[12] Ignatov, S. K.; Moltran v.2.5 - Program for molecular visualization and thermodynamic calculations. Uni. of Nizhny Novgorod, 2004.
[13] O’Boyle, N. M.; Tenderholt, A. L.; Langner, K. M.; “A library for package-independent computational chemistry algorithms”. J. Comput. Chem., 29(5): 839–845, 2008.
[14] William, H.; Andrew, D.; Klau, S.; VMD - Visual Molecular Dynamics, J. Mol. Graphics., 14: 33-38, 1996
[15] Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W.; “Revealing noncovalent interactions”. J. Am. Chem. Soc., 132(18): 6498-506, 2010
[16] Lu, T.; Chen, F.; “Multiwfn: A Multifunctional Wavefunction Analyser”. J. Comput. Chem. 33: 580-592, 2012
[17] Ylivainio, K. J.; Sufyan, A.; Larsson, J. A.; “A quantitative relationship between electron localization function and the strength of physical binding”. J. Phy.: Condens. Matter., 37(20); 205502, 2025.
[18] Kores, J. J.; Danish, I. A.; Sasitha, T.; Stuart, J. G.; Pushpam, E. J.; Jebaraj, J. W.; “Spectral, NBO, NLO, NCI, aromaticity and charge transfer analyses of anthracene-9, 10-dicarboxaldehyde by DFT”. Heliyon, 7(11), 2021.
[19] Murray, J. S.; Politzer, P.; “The electrostatic potential: an overview. Wiley Interdisciplinary Reviews”. Comput. Mol. Sci., 1(2): 153–163, 2011.
[20] Bishwokarma, N.; Budha, C.; Teemilsina, N. K.; Rai, K. B. “Exploring vibrational spectra, electronic properties and thermal analysis of Isoguanine molecule using DFT”. Scientific World, 18(18): 5–14, 2025.
[21] Assad, H.; Thakur, A.; Sharma, A. K.; Kumar, A.; “Density functional theory-based molecular modeling. In Computational Modelling and Simulations for Designing of Corrosion Inhibitors”. Elsevier, 95-113, 2023
[22] Contreras-García, J.; Johnson, E. R.; Keinan, S.; Chaudret, R.; Piquemal, J. P.; et al. “NCIPLOT: A Program for Plotting Noncovalent Interaction Regions”. J. Chem. Theory Comput., 7: 625-632, 2011.
[23] Johnson, E. R.; Keinan, S.; Mori Sanchez, P.; Contreras-Garcia, J.; Cohen, A. J.; et al.; “Revealing Noncovalent Interactions”. J. Am. Chem. Soc., 132: 6498-6506, 2010.
[24] Saleh, G.; Gatti, C.; Presti, L. L.; “Non-covalent interaction via the reduced density gradient: Independent atom model vs experimental multipolar electron densities”. Comput. Theor. Chem., 998: 148-163, 2012.
[25] Janani, S.; Rajagopal, H.; Muthu, S.; Aayisha, S.; Raja, M.; “Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR), HOMO-LUMO, chemical reactivity, AIM, ELF, LOL and Molecular docking studies on 1-Benzyl-4-(N-Boc-amino) piperidine” J. Mol. Struct., 1230: 129657, 2021
[26] Dhanalakshmi, E.; Rajesh, P.; Arunkumar, K.; Gnanasambandan, T.; Issaoui, N.; Sudha, K.; Raja, M.; “Synthesis, GCMS, spectroscopic, electronic properties, chemical reactivity, RDG, topology and biological assessment of 1-(3, 6, 6-trimethyl-1, 6, 7, 7a-tetrahydrocyclopenta [c] pyran-1-yl) ethenone”. Chem. Phys. Impact., 7: 100385, 2023.
[27] Gharti Magar, P.; Uprety, R.; Rai, K. B.; “First-principles DFT study of the molecular structure, spectroscopic analysis, electronic structures, and thermodynamic properties of ascorbic acid”. Himal. Phys., 11: 28-40, 2024.
[28] Yankova, R.; Genieva, S.; Halachev, N.; Dimitrova, G.; “Molecular structure, vibrational spectra, MEP, HOMO-LUMO and NBO analysis of Hf(SeO3)(SeO4)(H2O)4”. J. Mol. Struct., 1106: 82-88, 2016.
[29] Ojha, T.; Limbu, S.; Shrestha, P. M.; Gupta, S. P.; Rai, K. B.; “Comparative Computational Study on Molecular Structure, Electronic and Vibrational Analysis of Vinyl Bromide based on HF and DFT Approach”. Himal. J. Sci. Technol., 7(1): 38–49, 2023.
[30] Limbu, S.; Ojha, T.; Ghimire, R. R.; Rai, K. B.; “An investigation of vibrational analysis, thermodynamic properties, and electronic properties of Formaldehyde and its stretch by substituent acetone, acetyl chloride, and methyl acetate using first principles analysis”. BIBECHANA, 21(1): 23–36, 2024.
[31] Vincy, C. D.; Tarika, J. D.; Dexlin, X. D.; Rathika, A.; Beaula, T. J.; “Exploring the antibacterial activity of 1, 2-diaminoethane hexanedionic acid by spectroscopic, electronic, ELF, LOL, RDG analysis, and molecular docking studies using the DFT method”. J. Mol. Struct., 1247: 131388, 2022.
[32] Hajam, T. A.; Sallem, H.; Padhusha, M. S. A.; Mohammed Ameen, K. K.; “Synthesis, quantum chemical calculations and molecular docking studies of 2-ethoxy-4[(2-trifluoromethyl-phenylimino) methyl] phenol”. Mol. phys., 118(1):1-19, 2020.
[33] Mandal, S.; Reber, A. C.; Qian, M.; Weiss, P. S.; Khanna, S. N.; Sen, A.; “Controlling the band gap energy of cluster-assembled materials”. Acc. Chem. Res., 46(11): 2385-2395, 2013.
[34] Rai, K. B.; Ghimire, R. R.; Dhakal, C.; Pudasainee, K.; Siwakoti, B.; “Structural Equilibrium Configuration of Benzene and Aniline: A First-Principles Study”. J. Nepal Chem. Soc., 44(1): 1–15, 2024.
[35] Ahluwalia, V. K.; “Instrumental Methods of Chemical Analysis”. 1st ed.; Springer, Cham, Switzerland, 2023.
[36] Arivazhagan, M.; Jeyavijayan, S.; “Vibrational spectroscopic, first-order hyperpolarizability and HOMO, LUMO studies of 1, 2-dichloro-4-nitrobenzene based on Hartree–Fock and DFT calculations”. Spectrochim. Acta, Part A 79(2): 376-383, 2011.
[37] Sangeetha, T.; Dineshkumar, P.; Shanmugam, R.; Elangovan, A.; Arivazhagan, G.; “Hydrogen bond complexation of ethanol with chlorobenzene: FTIR studies”. Phys. Chem. Liq., 63(1): 1-12, 2025.
[38] Karaca, Ç.; “Spectroscopic (FT-Raman, FT-IR, UV-Vis, and NMR) and Theoretical Analysis of 1-Methylindole: Structural Characterization, Non-Covalent Interactions, and Electronic Properties”. Celal Bayar U. J. Sci., 21(1): 35-49, 2025.
[39] Faizan, M.; Bhat, S. A.; Alam, M. J.; Afroz, Z.; Ahmad, S.; “Anharmonic vibrational and electronic spectral study of 2-amino-4-hydroxy-6–methylpyrimidine: A combined experimental (FTIR, FT-Raman, UV–Vis) and theoretical (DFT, MP2) approach”. J. Mol. St., 1148: 89-100, 2017.
[40] Rijal, R.; Sah, M.; Lamichhane, H. P.; “Molecular simulation, vibrational spectroscopy, and global reactivity descriptors of pseudoephedrine molecule in different phases and states”. Heliyon, 9(3): e14801, 2023.
[41] El-Bindary, A. A.; Mohamed, G. G.; El-Sonbati, A. Z.; Diab, M. A.; Hassan, W. M. I.; Geometrical structure, potentiometric, “molecular docking and thermodynamic studies of azo dye ligand and its metal complexes”. J. Mol. Liq., 218: 138-149, 2016.
[42] Uprety, R.; Ghimire, R.; Gharti Magar, P.; Rokka, D.; Khadka, I. B.; Neupane., R.; Rai, K. B.; “Study of the molecular structure, electronic structure, spectroscopic analysis and thermodynamic properties of dibenzofuran using first principles”. J. Nepal Chem. Soc., 10(2): 8–18, 2024.
Downloads
Published
Issue
Section
License
Copyright (c) 2025 Tulsi Ojha, Prakash Man Shrestha, Suresh Prasad Gupta, Krishna Bahadur Rai

This work is licensed under a Creative Commons Attribution 4.0 International License.

.jpg)




